The AuReMe Solution
Current methods allow the reconstruction of high-quality GSMs but do not always take into account the need for metadata storage and exploitation to facilitate the study and reproducibility of models.
We designed a unified workspace AuReMe (AUtomated REconstruction of MEtabolic models), built around a Python package PADMet (Python library for hAndling metaData of METabolism), to house the reconstruction of GSMs.
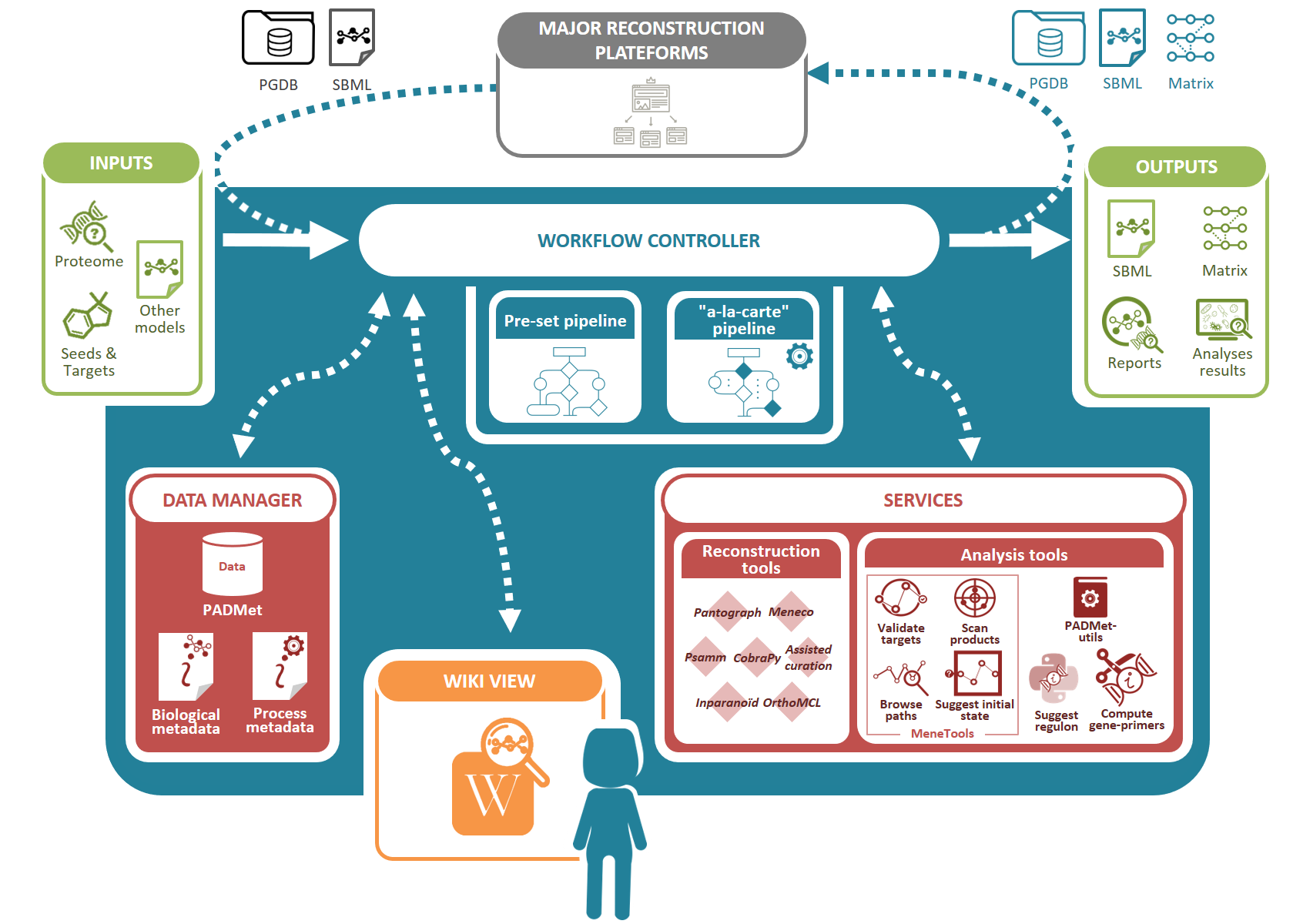
It gathers academic-free tools and enables the design of reconstruction pipelines that are flexible and can suit various available data sources (genome annotation, template GSMs, protein sequences, etc.) while storing metadata to ensure reproducibility of reconstructions.
It can follow four major steps of reconstruction processes:
- annotation-based modeling
- orthology-based modeling
- gap-filling
- manual curation
In addition, AuReMe supports most processes of the Thiele and Palsson protocol (Thiele & Palsson 2010) by proposing tools and methods that facilitate analysis and storing of the results at each step that is related to experiments or exploration of literature. In particular, the refinements to reconstruction box are strongly related to the management of metadata performed in AuReMe. Manual curation is assisted and formalized thanks to the PADMet package.
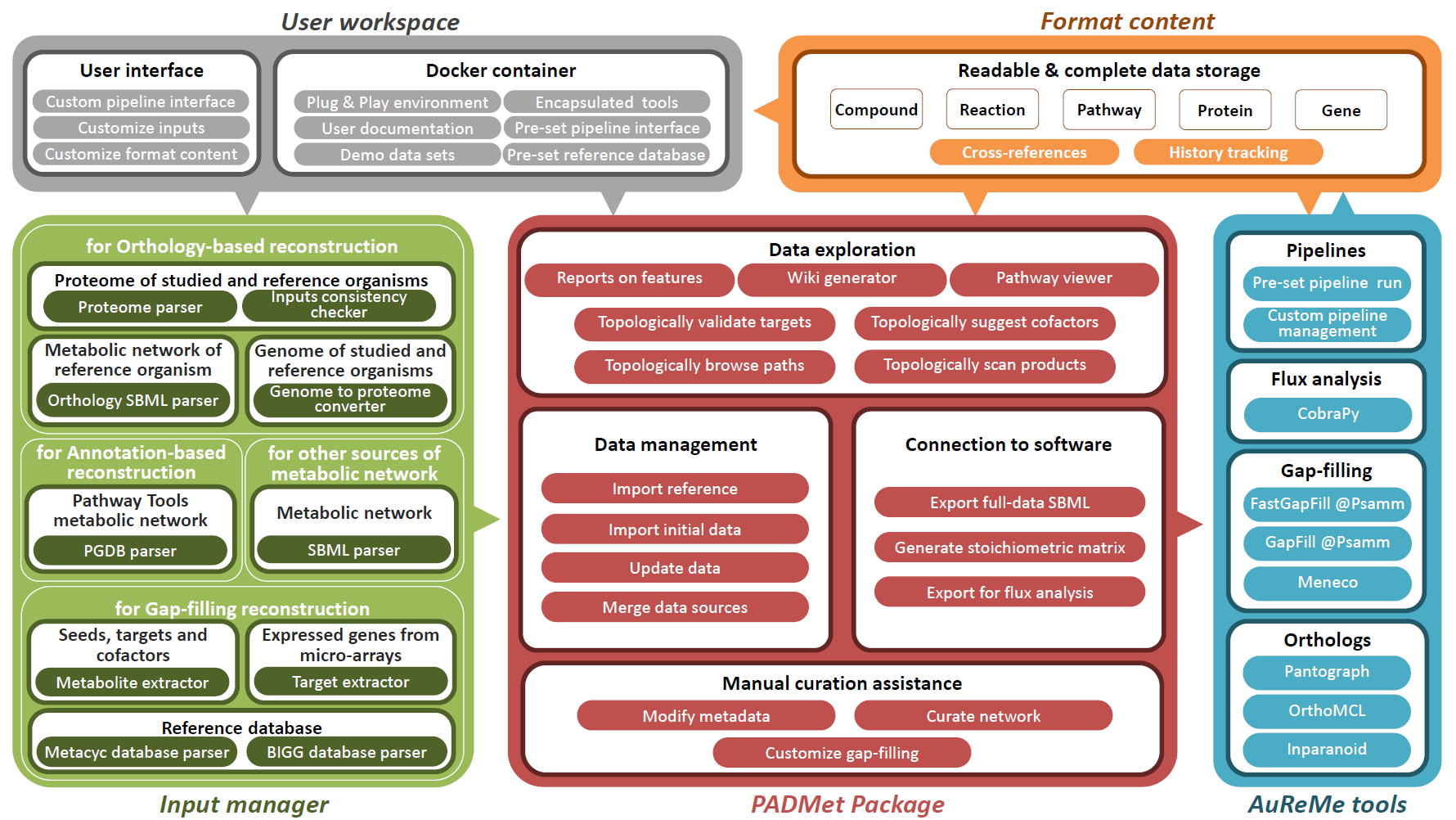
Additionally, analysis tools based on flux or topology are also included in the workspace. Contrary to existing platforms, AuReMe works with two major databases that are freely or academic-freely available: BiGG and MetaCyc, for which some versions are already included. The use of those databases facilitates the open-data initiative, that our workspace wants to promote. At any time of the reconstruction process, the visualization of model data and associated metadata is available through the generation of a local wiki, that can be connected to the MetaCyc or BiGG database used for reconstruction.
Accepted inputs are genome genbank files (gbk or gbff), protein sequence fasta files (faa), metabolic models Systems Biology Markup Language files (SBML (Hucka et al. 2003)) for studied or template organisms, Pathway/Genome Databases (PGDB) resulting from Pathway Tools workflows and text files for gene expression data, growth media composition, metabolic targets or biomass components. It is also possible to input a whole new database of reactions and metabolites in a tab-separated value format.
© 2013, creativeLabs. Bootstrap Themes Designed by BootstrapMaster in Poland 